Introduction
MGnifyR is a package designed to ease access to the
EBI’s MGnify resource,
allowing searching and retrieval of multiple datasets for downstream
analysis.
The latest version of MGnifyR seamlessly integrates with the miaverse framework providing access to cutting-edge tools in microbiome down-stream analytics.
Installation
MGnifyR is hosted on Bioconductor, and can be installed
using via BiocManager.
BiocManager::install("MGnifyR")Load MGnifyR package
Once installed, MGnifyR is made available in the usual
way.
library(MGnifyR)
#> Loading required package: MultiAssayExperiment
#> Loading required package: SummarizedExperiment
#> Loading required package: MatrixGenerics
#> Loading required package: matrixStats
#>
#> Attaching package: 'MatrixGenerics'
#> The following objects are masked from 'package:matrixStats':
#>
#> colAlls, colAnyNAs, colAnys, colAvgsPerRowSet, colCollapse,
#> colCounts, colCummaxs, colCummins, colCumprods, colCumsums,
#> colDiffs, colIQRDiffs, colIQRs, colLogSumExps, colMadDiffs,
#> colMads, colMaxs, colMeans2, colMedians, colMins, colOrderStats,
#> colProds, colQuantiles, colRanges, colRanks, colSdDiffs, colSds,
#> colSums2, colTabulates, colVarDiffs, colVars, colWeightedMads,
#> colWeightedMeans, colWeightedMedians, colWeightedSds,
#> colWeightedVars, rowAlls, rowAnyNAs, rowAnys, rowAvgsPerColSet,
#> rowCollapse, rowCounts, rowCummaxs, rowCummins, rowCumprods,
#> rowCumsums, rowDiffs, rowIQRDiffs, rowIQRs, rowLogSumExps,
#> rowMadDiffs, rowMads, rowMaxs, rowMeans2, rowMedians, rowMins,
#> rowOrderStats, rowProds, rowQuantiles, rowRanges, rowRanks,
#> rowSdDiffs, rowSds, rowSums2, rowTabulates, rowVarDiffs, rowVars,
#> rowWeightedMads, rowWeightedMeans, rowWeightedMedians,
#> rowWeightedSds, rowWeightedVars
#> Loading required package: GenomicRanges
#> Loading required package: stats4
#> Loading required package: BiocGenerics
#> Loading required package: generics
#>
#> Attaching package: 'generics'
#> The following objects are masked from 'package:base':
#>
#> as.difftime, as.factor, as.ordered, intersect, is.element, setdiff,
#> setequal, union
#>
#> Attaching package: 'BiocGenerics'
#> The following objects are masked from 'package:stats':
#>
#> IQR, mad, sd, var, xtabs
#> The following objects are masked from 'package:base':
#>
#> anyDuplicated, aperm, append, as.data.frame, basename, cbind,
#> colnames, dirname, do.call, duplicated, eval, evalq, Filter, Find,
#> get, grep, grepl, is.unsorted, lapply, Map, mapply, match, mget,
#> order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank,
#> rbind, Reduce, rownames, sapply, saveRDS, table, tapply, unique,
#> unsplit, which.max, which.min
#> Loading required package: S4Vectors
#>
#> Attaching package: 'S4Vectors'
#> The following object is masked from 'package:utils':
#>
#> findMatches
#> The following objects are masked from 'package:base':
#>
#> expand.grid, I, unname
#> Loading required package: IRanges
#> Loading required package: Seqinfo
#> Loading required package: Biobase
#> Welcome to Bioconductor
#>
#> Vignettes contain introductory material; view with
#> 'browseVignettes()'. To cite Bioconductor, see
#> 'citation("Biobase")', and for packages 'citation("pkgname")'.
#>
#> Attaching package: 'Biobase'
#> The following object is masked from 'package:MatrixGenerics':
#>
#> rowMedians
#> The following objects are masked from 'package:matrixStats':
#>
#> anyMissing, rowMedians
#> Loading required package: TreeSummarizedExperiment
#> Loading required package: SingleCellExperiment
#> Loading required package: Biostrings
#> Loading required package: XVector
#>
#> Attaching package: 'Biostrings'
#> The following object is masked from 'package:base':
#>
#> strsplitCreate a client
All functions in MGnifyR make use of a
MgnifyClient object to keep track of the JSONAPI url, disk
cache location and user access tokens. Thus the first thing to do when
starting any analysis is to instantiate this object. The following
snippet creates this.
mg <- MgnifyClient(useCache = TRUE)
mg
#> An object of class "MgnifyClient"
#> Slot "databaseUrl":
#> [1] "https://www.ebi.ac.uk/metagenomics/api/v1"
#>
#> Slot "authTok":
#> [1] NA
#>
#> Slot "useCache":
#> [1] TRUE
#>
#> Slot "cacheDir":
#> [1] "/tmp/Rtmpo4himI/.MGnifyR_cache"
#>
#> Slot "showWarnings":
#> [1] FALSE
#>
#> Slot "clearCache":
#> [1] FALSE
#>
#> Slot "verbose":
#> [1] TRUEThe MgnifyClient object contains slots for each of the
previously mentioned settings.
Functions for fetching the data
Search data
doQuery() function can be utilized to search results
such as samples and studies from MGnify database. Below, we fetch
information drinking water samples.
# Fetch studies
samples <- doQuery(
mg,
type = "samples",
biome_name = "root:Environmental:Aquatic:Freshwater:Drinking water",
max.hits = 10)The result is a table containing accession IDs and description – in this case – on samples.
Find relevent analyses accessions
Now we want to find analysis accessions. Each sample might have multiple analyses. Each analysis ID corresponds to a single run of a particular pipeline on a single sample in a single study.
analyses_accessions <- searchAnalysis(mg, "samples", samples$accession)By running the searchAnalysis() function, we get a
vector of analysis IDs of samples that we fed as an input.
analyses_accessions |> head()
#> [1] "MGYA00652201" "MGYA00652185" "MGYA00643487" "MGYA00643486" "MGYA00643485"
#> [6] "MGYA00643484"Fetch metadata
We can now check the metadata to get hint of what kind of data we
have. We use getMetadata() function to fetch data based on
analysis IDs.
analyses_metadata <- getMetadata(mg, analyses_accessions)The returned value is a data.frame that includes
metadata for example on how analysis was conducted and what kind of
samples were analyzed.
Fetch microbiome data
After we have selected the data to fetch, we can use
getResult()
The output is TreeSummarizedExperiment
(TreeSE) or MultiAssayExperiment
(MAE) depending on the dataset. If the dataset includes
only taxonomic profiling data, the output is a single
TreeSE. If dataset includes also functional data, the
output is multiple TreeSE objects that are linked together
by utilizing MAE.
mae <- getResult(mg, accession = analyses_accessions)
mae
#> A MultiAssayExperiment object of 6 listed
#> experiments with user-defined names and respective classes.
#> Containing an ExperimentList class object of length 6:
#> [1] microbiota: TreeSummarizedExperiment with 3506 rows and 50 columns
#> [2] go-slim: TreeSummarizedExperiment with 116 rows and 38 columns
#> [3] go-terms: TreeSummarizedExperiment with 3133 rows and 38 columns
#> [4] interpro-identifiers: TreeSummarizedExperiment with 18223 rows and 38 columns
#> [5] taxonomy: TreeSummarizedExperiment with 3617 rows and 50 columns
#> [6] taxonomy-lsu: TreeSummarizedExperiment with 3378 rows and 42 columns
#> Functionality:
#> experiments() - obtain the ExperimentList instance
#> colData() - the primary/phenotype DataFrame
#> sampleMap() - the sample coordination DataFrame
#> `$`, `[`, `[[` - extract colData columns, subset, or experiment
#> *Format() - convert into a long or wide DataFrame
#> assays() - convert ExperimentList to a SimpleList of matrices
#> exportClass() - save data to flat filesYou can get access to individual TreeSE object in
MAE by specifying index or name.
mae[[1]]
#> class: TreeSummarizedExperiment
#> dim: 3506 50
#> metadata(0):
#> assays(1): counts
#> rownames(3506): 82608 62797 ... 5820 6794
#> rowData names(9): Kingdom Phylum ... taxonomy1 taxonomy
#> colnames(50): MGYA00144458 MGYA00144419 ... MGYA00652185 MGYA00652201
#> colData names(64): analysis_analysis.status analysis_pipeline.version
#> ... sample_geo.loc.name sample_instrument.model
#> reducedDimNames(0):
#> mainExpName: NULL
#> altExpNames(0):
#> rowLinks: NULL
#> rowTree: NULL
#> colLinks: NULL
#> colTree: NULLTreeSE object is uniquely positioned to support
SummarizedExperiment-based microbiome data manipulation and
visualization. Moreover, it enables access to miaverse
tools. For example, we can estimate diversity of samples…
library(mia)
#> This is mia version 1.19.2
#> - Online documentation and vignettes: https://microbiome.github.io/mia/
#> - Online book 'Orchestrating Microbiome Analysis (OMA)': https://microbiome.github.io/OMA/docs/devel/
mae[[1]] <- estimateDiversity(mae[[1]], index = "shannon")
#> Warning in estimateDiversity(mae[[1]], index = "shannon"): 'estimateDiversity'
#> is deprecated. Use 'addAlpha' instead.
library(scater)
#> Loading required package: scuttle
#> Loading required package: ggplot2
plotColData(mae[[1]], "shannon", x = "sample_environment..biome.")
… and plot abundances of most abundant phyla.
# Agglomerate data
altExps(mae[[1]]) <- splitByRanks(mae[[1]])
library(miaViz)
#> Loading required package: ggraph
#>
#> Attaching package: 'miaViz'
#> The following object is masked from 'package:mia':
#>
#> plotNMDS
# Plot top taxa
top_taxa <- getTopFeatures(altExp(mae[[1]], "Phylum"), 10)
#> Warning in getTopFeatures(altExp(mae[[1]], "Phylum"), 10): 'getTopFeatures' is
#> deprecated. Use 'getTop' instead.
plotAbundance(
altExp(mae[[1]], "Phylum")[top_taxa, ],
rank = "Phylum",
as.relative = TRUE
)
#> Warning: The following values are already present in `metadata` and will be
#> overwritten: 'agglomerated_by_rank'. Consider using the 'name' argument to
#> specify alternative names.
#> Warning: Removed 107 rows containing missing values or values outside the scale range
#> (`geom_bar()`).
We can also perform other analyses such as principal component analysis to microbial profiling data by utilizing miaverse tools.
# Apply relative transformation
mae[[1]] <- transformAssay(mae[[1]], method = "relabundance")
# Perform PCoA
mae[[1]] <- runMDS(
mae[[1]], assay.type = "relabundance",
FUN = vegan::vegdist, method = "bray")
# Plot
plotReducedDim(
mae[[1]], "MDS", colour_by = "sample_environment..biome.")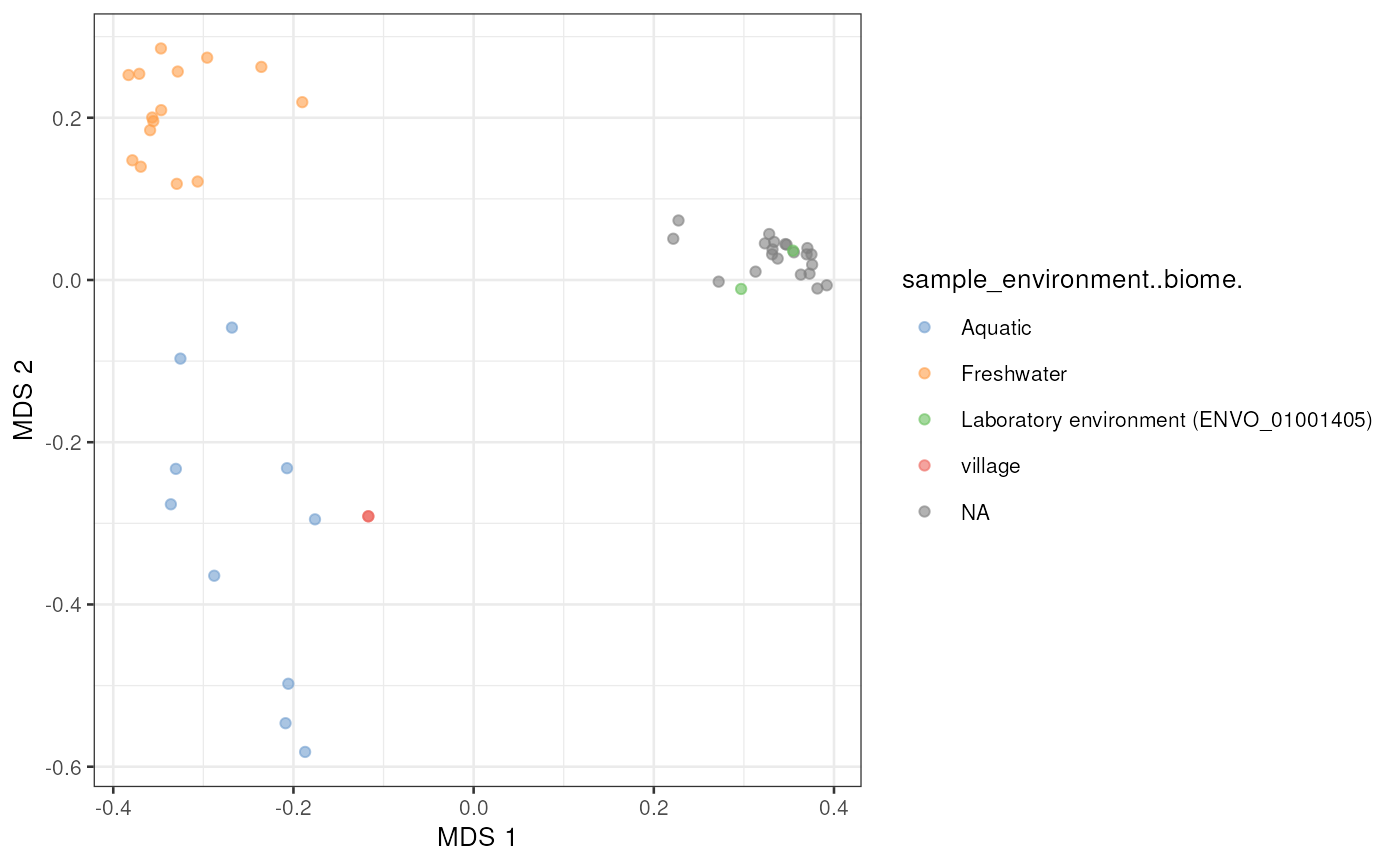
Fetch raw files
While getResult() can be utilized to retrieve microbial
profiling data, getData() can be used more flexibly to
retrieve any kind of data from the database. It returns data as simple
data.frame or list format.
publications <- getData(mg, type = "publications")
colnames(publications) |> head()
#> [1] "document.id" "type"
#> [3] "id" "attributes.pubmed-id"
#> [5] "attributes.pubmed-central-id" "attributes.pub-title"The result is a data.frame by default. In this case, it
includes information on publications fetched from the data portal.
Fetch sequence files
Finally, we can use searchFile() and
getFile() to retrieve other MGnify pipeline outputs such as
merged sequence reads, assembled contigs, and details of the functional
analyses.
With searchFile(), we can search files from the
database.
dl_urls <- searchFile(mg, analyses_accessions, type = "analyses")The returned table contains search results related to analyses that we fed as an input. The table contains information on file and also URL address from where the file can be loaded.
target_urls <- dl_urls[
dl_urls$attributes.description.label == "Predicted alpha tmRNA", ]
colnames(target_urls) |> head()
#> [1] "type" "id"
#> [3] "attributes.alias" "attributes.file.format.name"
#> [5] "attributes.file.format.extension" "attributes.file.format.compression"Finally, we can download the files with getFile().
# Just select a single file from the target_urls list for demonstration.
file_url <- target_urls$download_url[[1]]
cached_location <- getFile(mg, file_url)The function returns a path where the file is stored.
# Where are the files?
cached_location
#> [1] "/.MGnifyR_cache/analyses/MGYA00652201/file/ERZ20300939_alpha_tmRNA.RF01849.fasta.gz"
sessionInfo()
#> R Under development (unstable) (2026-02-08 r89382)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.3 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
#> [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
#> [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
#> [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
#> [9] LC_ADDRESS=C LC_TELEPHONE=C
#> [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats4 stats graphics grDevices utils datasets methods
#> [8] base
#>
#> other attached packages:
#> [1] miaViz_1.19.1 ggraph_2.2.2
#> [3] scater_1.39.2 ggplot2_4.0.2
#> [5] scuttle_1.21.0 mia_1.19.2
#> [7] MGnifyR_1.7.2 TreeSummarizedExperiment_2.19.0
#> [9] Biostrings_2.79.4 XVector_0.51.0
#> [11] SingleCellExperiment_1.33.0 MultiAssayExperiment_1.37.2
#> [13] SummarizedExperiment_1.41.1 Biobase_2.71.0
#> [15] GenomicRanges_1.63.1 Seqinfo_1.1.0
#> [17] IRanges_2.45.0 S4Vectors_0.49.0
#> [19] BiocGenerics_0.57.0 generics_0.1.4
#> [21] MatrixGenerics_1.23.0 matrixStats_1.5.0
#> [23] knitr_1.51 BiocStyle_2.39.0
#>
#> loaded via a namespace (and not attached):
#> [1] RColorBrewer_1.1-3 jsonlite_2.0.0
#> [3] magrittr_2.0.4 ggbeeswarm_0.7.3
#> [5] farver_2.1.2 rmarkdown_2.30
#> [7] fs_1.6.6 ragg_1.5.0
#> [9] vctrs_0.7.1 memoise_2.0.1
#> [11] DelayedMatrixStats_1.33.0 ggtree_4.1.1
#> [13] htmltools_0.5.9 S4Arrays_1.11.1
#> [15] BiocBaseUtils_1.13.0 BiocNeighbors_2.5.3
#> [17] janeaustenr_1.0.0 gridGraphics_0.5-1
#> [19] SparseArray_1.11.10 sass_0.4.10
#> [21] bslib_0.10.0 tokenizers_0.3.0
#> [23] htmlwidgets_1.6.4 desc_1.4.3
#> [25] plyr_1.8.9 DECIPHER_3.7.0
#> [27] cachem_1.1.0 igraph_2.2.2
#> [29] lifecycle_1.0.5 pkgconfig_2.0.3
#> [31] rsvd_1.0.5 Matrix_1.7-4
#> [33] R6_2.6.1 fastmap_1.2.0
#> [35] tidytext_0.4.3 aplot_0.2.9
#> [37] digest_0.6.39 ggnewscale_0.5.2
#> [39] patchwork_1.3.2 irlba_2.3.7
#> [41] SnowballC_0.7.1 textshaping_1.0.4
#> [43] vegan_2.7-2 beachmat_2.27.2
#> [45] labeling_0.4.3 urltools_1.7.3.1
#> [47] httr_1.4.7 polyclip_1.10-7
#> [49] abind_1.4-8 mgcv_1.9-4
#> [51] compiler_4.6.0 fontquiver_0.2.1
#> [53] withr_3.0.2 S7_0.2.1
#> [55] BiocParallel_1.45.0 viridis_0.6.5
#> [57] DBI_1.2.3 ggforce_0.5.0
#> [59] MASS_7.3-65 rappdirs_0.3.4
#> [61] DelayedArray_0.37.0 bluster_1.21.0
#> [63] permute_0.9-10 tools_4.6.0
#> [65] vipor_0.4.7 otel_0.2.0
#> [67] beeswarm_0.4.0 ape_5.8-1
#> [69] glue_1.8.0 nlme_3.1-168
#> [71] grid_4.6.0 cluster_2.1.8.2
#> [73] reshape2_1.4.5 gtable_0.3.6
#> [75] tidyr_1.3.2 BiocSingular_1.27.1
#> [77] tidygraph_1.3.1 ScaledMatrix_1.19.0
#> [79] ggrepel_0.9.6 pillar_1.11.1
#> [81] stringr_1.6.0 yulab.utils_0.2.4
#> [83] splines_4.6.0 dplyr_1.2.0
#> [85] tweenr_2.0.3 treeio_1.35.0
#> [87] lattice_0.22-9 tidyselect_1.2.1
#> [89] DirichletMultinomial_1.53.0 fontLiberation_0.1.0
#> [91] fontBitstreamVera_0.1.1 gridExtra_2.3
#> [93] bookdown_0.46 xfun_0.56
#> [95] graphlayouts_1.2.2 stringi_1.8.7
#> [97] ggfun_0.2.0 lazyeval_0.2.2
#> [99] yaml_2.3.12 evaluate_1.0.5
#> [101] codetools_0.2-20 gdtools_0.5.0
#> [103] tibble_3.3.1 BiocManager_1.30.27
#> [105] ggplotify_0.1.3 cli_3.6.5
#> [107] systemfonts_1.3.1 jquerylib_0.1.4
#> [109] Rcpp_1.1.1 triebeard_0.4.1
#> [111] parallel_4.6.0 pkgdown_2.2.0
#> [113] ecodive_2.2.2 sparseMatrixStats_1.23.0
#> [115] decontam_1.31.0 viridisLite_0.4.3
#> [117] tidytree_0.4.7 ggiraph_0.9.4
#> [119] scales_1.4.0 purrr_1.2.1
#> [121] crayon_1.5.3 rlang_1.1.7